Biologia Molecular
Medicina Preventiva
Confira os painéis que a Neurogene Laboratório
oferece em parceria com a FullDNA:
SAÚDE DA MULHER
Número de genes analisados: 802
Número de variantes analisadas: 1801
![]()
NUTRIÇÃO FUNCIONAL
Número de genes analisados: 1069
Número de variantes analisadas: 3051
![]()
LONGEVIDADE SAUDÁVEL
Número de genes analisados: 946
Número de variantes analisadas: 1982
![]()
HARMONIZAÇÃO FACIAL FEMININO
Número de genes analisados: 54
Número de variantes analisadas: 74
Número de Categorias: 1
Número de Condições: 21
![]()
HARMONIZAÇÃO FACIAL MASCULINO
Número de genes analisados: 54
Número de variantes analisadas: 74
Número de Categorias: 1
Número de Condições: 21
![]()
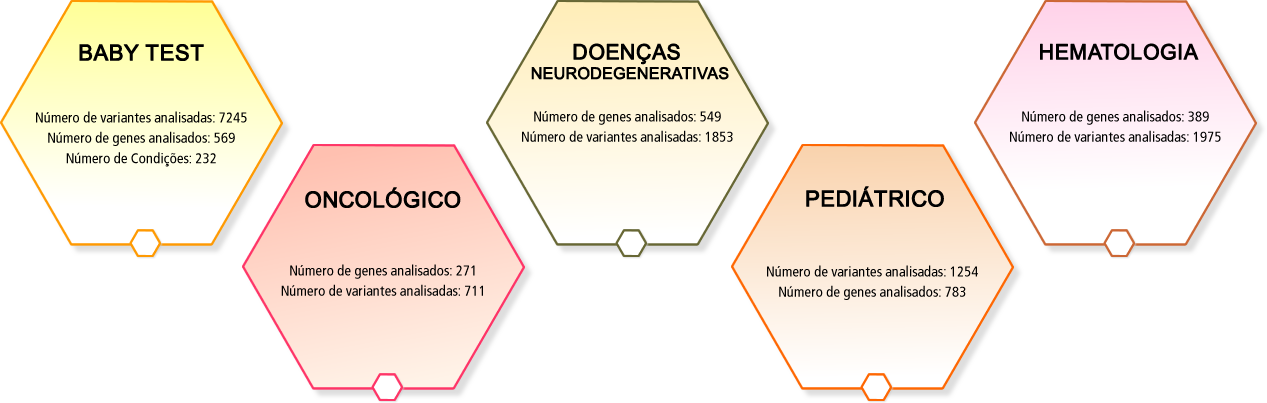
BABY TEST
Número de variantes analisadas: 7245
Número de genes analisados: 569
Número de Condições: 232
![]()
ONCOLÓGICO
Número de genes analisados: 271
Número de variantes analisadas: 711
![]()
DOENÇAS NEURODEGENERATIVAS
Número de genes analisados: 549
Número de variantes analisadas: 1853
![]()
PEDIÁTRICO
Número de variantes analisadas: 1254
Número de genes analisados: 783
![]()
HEMATOLOGIA
Número de genes analisados: 389
Número de variantes analisadas: 1975
![]()
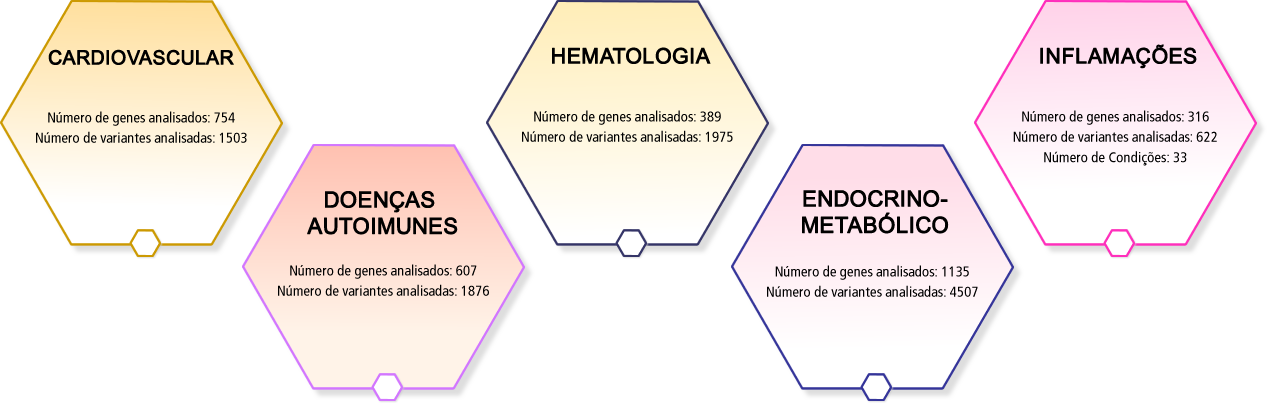
CARDIOVASCULAR
Número de genes analisados: 754
Número de variantes analisadas: 1503
![]()
DOENÇAS AUTOIMUNES
Número de genes analisados: 607
Número de variantes analisadas: 1876
![]()
HEMATOLOGIA
Número de genes analisados: 389
Número de variantes analisadas: 1975
![]()
ENDOCRINO-METABÓLICO
Número de genes analisados: 1135
Número de variantes analisadas: 4507
![]()
INFLAMAÇÕES
Número de genes analisados: 316
Número de variantes analisadas: 622
Número de Condições: 33
![]()
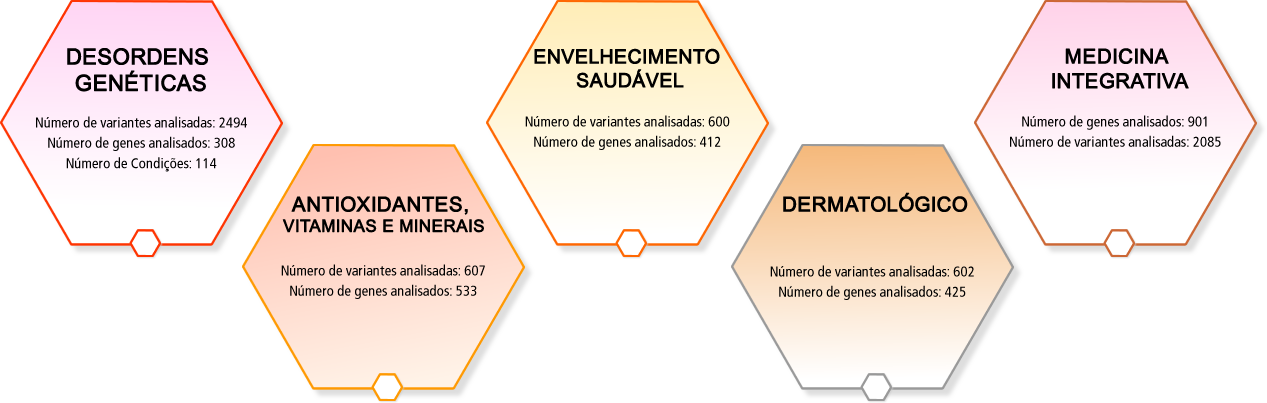
DESORDENS GENÉTICAS
Número de variantes analisadas: 2494
Número de genes analisados: 308
Número de Condições: 114
![]()
ANTIOXIDANTES, VITAMINAS E MINERAIS
Número de variantes analisadas: 607
Número de genes analisados: 533
![]()
ENVELHECIMENTO SAUDÁVEL
Número de variantes analisadas: 600
Número de genes analisados: 412
![]()
DERMATOLÓGICO
Número de variantes analisadas: 602
Número de genes analisados: 425
![]()
MEDICINA INTEGRATIVA
Número de genes analisados: 901
Número de variantes analisadas: 2085
Medicina Preventiva
Confira os painéis que a Neurogene Laboratório
oferece em parceria com a FullDNA:
Antígeno HLA-B27(Doença reumática crônica)
Os antígenos HLA são produtos dos genes do complexo maior de histocompatibilidade. Alguns destes antígenos estão relacionados a presença de determinadas doenças. A associação mais freqüente e a da Espondilite Anquilosante (EA) com o antígeno HLA-B27, que está presente em mais de 90% dos indivíduos de raça branca acometidos por essa doença.
A Espondilite Anquilosante (EA) é uma doença reumática inflamatória crônica que afeta a coluna vertebral, articulações, podendo ocorrer manifestações extra-esqueléticas como distúrbios cardiovasculares e visuais. A EA faz parte de um grupo de doenças conhecidas como espondiloartropatias e se relacionam pela ocorrência do marcador HLA-B27.A pesquisa também e indicada para identificar risco do acometimento de descendentes. Elevada incidência do antígeno HLA-B27 tem sido relatada na síndrome de Reiter, uveite anterior, artrite reativa e artrite psoriatica. Este antígeno não e um marcador da doença, uma vez que esta presente em aproximadamente 10% dos indivíduos normais. O resultado deve ser associado aos achados clínicos e radiológicos sugestivos destas doenças. As vantagens da PCR sobre a citometria de fluxo incluem interpretação objetiva e maior especificidade (a citometria de fluxo pode apresentar reação cruzada com o HLA-B7, HLA-B37 e HLA-B39).
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Defeitos no tubo neural (PCR MTHFR)
Os defeitos do tubo neural (DTN) são malformações congênitas que ocorrem durante o período de formação do embrião.
Os DTN incluem problemas de espinha bífida (insuficiência da fusão dos arcos vertebrais que circundam a medula espinhal) e anencefalia (malformação congênita em que não há formação de todo o cérebro ou de parte dele). Tratam-se de defeitos congênitos relativamente comuns. Por isso aconselha-se que, ao engravidar, a mulher esteja tomando ácido fólico.
O governo americano determinou, em 1988, que várias classes de alimentos passassem a ser enriquecidas com ácido fólico, ocasionando uma queda de 19% no número de bebês nascidos com espinha bífida e anencefalia.
O ácido fólico pode ser encontrado naturalmente em alimentos como fígado, espinafre e brócolis. Na gestação há necessidade de dobrar o consumo da substância ou de haver complementação alimentar.
Indicação:
O Exame de Prevenção a Defeitos do Tubo Neural, realizado pelo Laboratório Neurogene, é indicado para os seguintes casos:
1) Mulheres que tiveram filhos com Defeitos do Tubo Neural e que desejem dar à luz novamente;
2) Mulheres cujas mães tiveram filhos com DTN e que pretendam engravidar.
Nota: Este exame é realizado uma única vez na vida, em qualquer idade, inclusive pré-natal, pois as informações estudadas são as do código genético, o qual não sofre alterações depois de formado.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Hemocromatose hereditária
A hemocromatose hereditária (HH) é a mais comum doença genética, hereditária, na população caucasiana (branca), alcançando até 1 em 200 pessoas descendentes de nórdicos ou celtas. Trata-se de uma predisposição para a absorção excessiva de ferro da alimentação. Este ferro acumula-se principalmente no fígado, pâncreas e coração, podendo levar ao óbito por cirrose, hepatocarcinoma, insuficiência cardíaca ou diabetes.
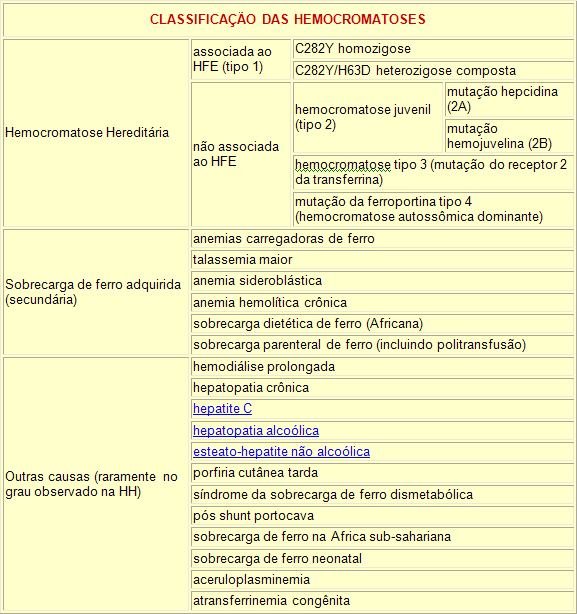
O controle da quantidade de ferro em nosso organismo é basicamente dependente da absorção intestinal do mesmo. Não temos nenhum mecanismo fisiológico para eliminar o ferro se ele estiver em excesso, além de controlar a absorção e depender da perda mínima decorrente da perda de células, como células intestinais e sangue durante a menstruação.
Na hemocromatose hereditária, mutações genéticas (geralmente transmitidas de pais para filhos) levam a um aumento na absorção do ferro no intestino (duodeno e jejuno proximal), o que leva ao acúmulo do metal no organismo.
A Hemocromatose Hereditária é caracterizada por dois pontos de mutação no gene localizado no cromossomo 6: C282Y e H63D. A mutação C282Y é mais significante clinicamente do que a H63D, pois ela ocorre em 85 % dos pacientes com HH. Nessa mutação, ocorre a substituição de cisteina por tirosina no aminoácido 282 da proteína HFE.
A mutação H63D é menos significante clinicamente, a menos que esteja associada à heterozigose para mutação C282Y ( C282Y / H63D). A proteína HFE resultante dessa mutação exibe a substituição do aminoácido histidina pelo ácido aspártico na posição 63 ( H63D).
Referência: Feder, J.N, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature, 13: 399-408, 1996.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Infertilidade masculina
A esterilidade masculina por azoospermia ou oligospermia severa afeta aproximadamente 7-10% de todos os homens. Afastadas causas obstrutivas, a etiologia mais comum da azoospermia é genética: 10-15% dos pacientes apresentam alterações dos cromossomos sexuais (Síndrome de Klinefelter – 47,XXY, ou 46,XX) e outros 10-20% dos pacientes apresentam microdeleções do braço longo do cromossomo Y, que só podem ser diagnosticadas por estudos moleculares. As microdeleções ocorrem em uma região do cromossomo Y chamada AZF, onde concentram-se vários genes envolvidos na espermatogênese. O diagnóstico exato da causa da azoospermia é muito importante para orientação do tratamento do casal infértil.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
PESQUISA DO GENE SRY POR PCR
O gene SRY é responsável pelo identificação do cromossomo Y, que determina o gênero masculino. Este exame é de vital importância na detecção de síndrome de Turner, nascimento de crianças com genitália ambígua. A definição do sexo é importante para se registrar a criança e definir socialmente seu sexo genético.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Leucemias
PCR PARA TRANLOCAÇÃO BCR/ABL – CROMOSSOMO PHILADELPHIA
A leucemia mielóide crônica se caracteriza na maioria dos casos, pela presença de uma anormalidade genética que foi chamada de cromossomo Philadelphia. O cromossomo Philadelphia é uma anormalidade que envolve os cromossomos de números 9 e 22. Esses cromossomos se quebram e trocam partes entre si. Esta alteração é chamada translocação e o novo cromossomo que se forma é denominado Philadelphia ou translocação t(9:22).
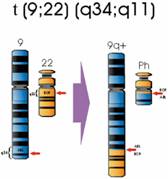
Fig. 1 Cromossomo Philadelphia (Ph). Translocação recíproca entre os cromossomos 9 e 22; t (9;22) (q34;q11).
Método: O teste bcr/abl pela técnica RT-PCR (Reação em Cadeia da Polimerase com transcrição reversa) em RNA extraído de sangue ou medula óssea, é qualitativo e permite o diagnóstico de certeza da presença – ou da ausência – do transcrito quimérico resultante da translocação recíproca entre os cromossomos 9; 22 que origina o cromossomo Philadelphia.
Material: 5ml de Sangue com EDTA ou 3ml de Medula Óssea com EDTA.
Prazo de Entrega: 20 dias úteis
PCR PARA TRANSLOCAÇÃO 15;17 (PML-RARA)
Leucemia Promielocítica Aguda (LPA), representa 5 a 8% das Leucemias Mielóides Agudas e ocorre em qualquer faixa etária e está associada a coagulação intravascular disseminada. A translocação t(15;17) envolvendo os protooncogenes PML (promielocítico) e RAR nos cromossomos 15q e 17q, respectivamente, presentes na Leucemia Mielóide Aguda é do tipo M3.
Método: No Laboratório Neurogene, esse rearranjo é detectado por técnica de RT-PCR. O teste é feito pela técnica RT-PCR (Reação em Cadeia da Polimerase com transcrição reversa) em RNA extraído de sangue ou medula óssea, é qualitativo e permite o diagnóstico da presença – ou da ausência da translocação 15;17.
Material: 5ml de Sangue com EDTA ou 3ml de Medula Óssea com EDTA.
Prazo de Entrega: 30 dias
Osteogênesis imperfeita (Gene COL Ia1)
Osteogenesis Imperfecta (OI) é um grupo de condições heterogêneas que se caracteriza por acentuada fragilidade óssea. Na grande maioria dos casos, mutações no gene do colágeno tipo 1α1(COLIA1) e α2 (COLIA2) são responsáveis pelo fenótipo; no entanto, mutações em outros locis genéticos podem produzir fenótipos semelhantes.
A característica primordial da OI é a fragilidade óssea, todas as outras características são variáveis com heterogeneidade clínica em diferentes membros de uma mesma família.
A prevalência da OI é estimada em 1:15000-20000 crianças (2).
O defeito básico dessa patologia é a osteopenia que pode ser acompanhado de fraqueza muscular e frouxidão ligamentar. As crianças afetadas podem apresentar fraturas recorrentes que levam a imobilização e a dor principalmente na idade pré-escolar. Essa fragilidade de uma maneira geral persiste por toda vida comprometendo a qualidade de vida do paciente.
Na última década a concepção da OI tem mudado, passou de uma desordem causada por mutação do colágeno, dividida em quatro tipos, para a qual não havia tratamento clínico para grupo mais abrangente, causada por outras mutações além do COLIA e com possibilidade terapêutica sintomática eficaz.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Pesquisa genética para Doença Celíaca
A doença celiaca é uma enteropatia imunologicamente mediada e desencadeada por ingestão de glúten em indivíduos geneticamente suscetiveis, com prevalência estimada de 1/300.
Por conta de sua clínica heterogênea, o diagnostico da moléstia depende muitas vezes de um abordagem clínica, laboratorial e histopatológica combinada. Recentemente, demonstrou-se que a presença dos marcadores DQ2 e DQ8, do grupo HLA-DQ, que agora são pesquisados pelo Laboratório Neurogene, está associada à doença celíaca.
Em pacientes caucasóides, cerca de 90% possuem o DQ2 e a maior parte dos 10% restantes apresenta o DQ8. Por outro lado, os alelos que codificam as moléculas DQ2 e DQ8 são encontrados em cerca de 15% a 30% da população geral, razão pela qual sua presença não tem especificidade para detectar a doença celíaca.
O exame no entanto, ajuda a excluir essa possibilidade, já que a ausência de ambos os marcadores torna a hipótese de um indivíduo ser portador da doença extremamente improvável. Aliado à pesquisa de anticorpos antiendomísio e antitransglutaminase, esse teste também pode ser útil na investigação de casos que apresentem biópsia de jejuno inconclusiva e na abordagem dos pacientes com restrições à realização da biópsia.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Predisposição à hipertensão arterial e doenças cardiovasculares
Síndrome de Prader Willi e Angelman
A Síndrome de Prader-Willi e a Síndrome de Angelman cada uma ocorrem em uma freqüência de cerca de 1/10.000 a 1/20.000 nascidos. A razão pela qual elas são discutidas juntas é que ambas síndromes são mais frequentemente causadas por microdeleções na região cromossômica 15q11-q13. A SPW e SA são associadas com deficiências do cromossomo 15 paterno e materno, respectivamente. As deficiências são causadas por microdeleções (~75 % de casos de SPW ou SA), por dissomia uniparental
(25 % de SPW e 2 % de SA) ou por mutações na impressão genômica (Kosaki, K. et al 2002).
Método: É utilizado o método de PCR específico para amplificação das cópias metilada e não metilada do exon a do gene SNRPN, para diagnóstico da SPW e SA.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 15 dias úteis
Síndrome de Turner
A Síndrome de Turner é uma das anormalidades genéticas mais comuns. Acomete 1 em cada 2.000 nascidos vivos do sexo feminino. As manifestações clínicas mais freqüentes são baixa estatura, disgenesia gonadal, malformação renal e anormalidades cardiovasculares. Esta síndrome é caracterizada por monossomia do cromossomo X (45,X), presente em 50 a 60 % dos casos, ao passo que o restante dos pacientes tem uma grande variabilidade de anomalias do cromossomo X, incluindo vários mosaicismos.
Cromossomo Y
Dos pacientes estudados por citogenética, 6% apresentam o cromossomo Y ou resíduos dele, sendo que em outros 3% só se encontra este cromossomo através da técnica de PCR. A presença deste cromossomo, ou parte dele, tem uma forte associação com o risco (> 35%) de desenvolvimento do gonadoblastoma, que corresponde a um tumor do ovário de células indiferenciadas. Isso justifica a necessidade de identificar os pacientes de risco a fim de estabelecer medidas médicas preventivas, como gonadectomia (retirada dos ovários em fita) ou avaliação da gônada por via laparoscópica. Os estudos na área de biologia molecular possibilitaram o desenvolvimento de técnicas bastante sensíveis para detectar o cromossomo Y no DNA dos pacientes. Usando a técnica de PCR (Reação em Cadeia da Polimerase) e sondas específicas, é possível identificar a presença do gene determinante do sexo no cromossomo Y – SRY, o gene da proteína específica testicular – TSPY e outras seqüências gênicas presentes no cromossomo Y, como DAZ1. A utilização concomitante destas três sondas aumenta a sensibilidade do método e garante a detecção de baixos níveis de mosaicismo. Este estudo tem esta característica interessante, pois, através da biologia molecular, pode-se prevenir o desenvolvimento de um câncer.
O estudo destes marcadores do cromossomo Y (SRY, TSPY, DAZ1) está indicado para todos os pacientes com diagnóstico de Síndrome de Turner. O material utilizado é 2 mL de sangue em EDTA, em temperatura ambiente.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes SRY, TSPY e DAZ-1.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 15 dias úteis
Síndrome do X Frágil
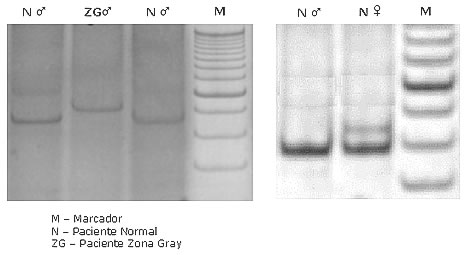
A Síndrome do X Frágil é causada pela expansão de uma seqüência de trinucleotídeos (CGG) localizada na região 5’UTR do gene FMR1 (Fragile X Mental Retardation), situado no cromossomo X (Xq27.3). Dependendo do número de repetições CGG, os indivíduos podem ser classificados em 4 categorias: normais (apresentam de 6 a 40 repetições CGG), zona gray (41 a 60), pré-mutados (61 a 200) e afetados (mais de 200). Indivíduos afetados, além de possuírem grande número de repetições CGG, normalmente apresentam metilação neste mesmo gene, resultando redução da expressão da proteína FMRP, codificada pelo gene. A ausência da FMRP está fortemente associada ao retardo mental apresentado pelos portadores da Síndrome do X Frágil, que possuem de 0 a 15% de FMRP em linfócitos, enquanto pessoas normais apresentam de 80 a 100%.
Desde o ano 2000 o Laboratório Neurogene investe no diagnóstico da Síndrome do X Frágil, nossa intenção é o destaque e reconhecimento como laboratório de referência nesta área. Disponibilizamos aos nossos laboratórios conveniados técnicas pioneiras para o estudo desta síndrome, aos nossos pacientes, oferecemos a continuidade da investigação familiar juntamente com o aconselhamento genético. Além disso, o Laboratório Neurogene participa voluntariamente de várias atividades vinculadas a Associação Catarinense do X Frágil, oferecendo palestras esclarecedoras sobre a síndrome para médicos, escolas, pais e demais interessados.
Método: Estudo realizado através da amplificação da zona repetitiva (CGG)n do gene FMR-1, utilizando a técnica da PCR (par de primers específicos para a mutação FRAXA). Teste de triagem, permite a visualização de repetições CGG de pacientes normais e zona gray de pequena expansão (até 50 CGG). Técnica eficiente no diagnóstico de pacientes do sexo masculino e em algumas pacientes do sexo feminino heterozigotas.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 20 dias
Técnica específica para detecção até 150 repetições de CGG
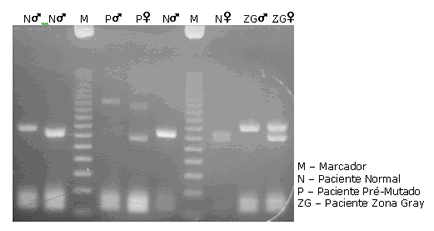
Método: Estudo realizado através da amplificação da zona repetitiva (CGG)n do gene FMR-1, utilizando a técnica da PCR (par de primers específicos para a mutação FRAXA e Expand Long Template PCR System). Fornece o tamanho aproximado dos alelos de pacientes normais, zona gray e pré-mutados. Técnica também eficiente na definição do diagnóstico de mulheres heterozigotas, em homozigotas é possível definir diagnóstico por estudo materno e paterno.
O Laboratório Neurogene é pioneiro neste tipo de análise, graças à uma relação de parceria com a Universidade de Davis, Califórnia, EUA.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
(Exame terceirizado, em breve será realizado pelo Laboratório Neurogene)
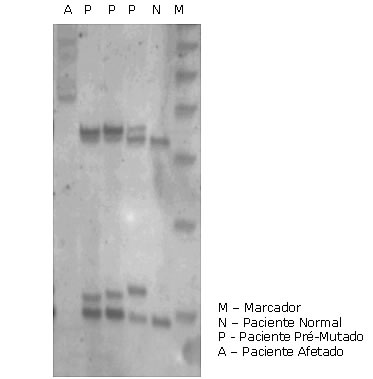
Método: O DNA genômico é isolado, digerido com enzimas de restrição (EcoRI e NruI) e transferidos para membrana de nylon, que é hibridizada com a Sonda StB12.3 marcada com Dig-11-dUTP e exposta a raio X por duas horas. Permite a visualização do número de repetições CGG em Kb e informa o grau de metilação do gene, eficaz no diagnóstico de afetados, mutação completa.
Material: 10ml de sangue com EDTA
Prazo de Entrega: 90 dias
SOUTHERN BLOTTING PARA X FRÁGIL
Surdez neurossensorial não sindrômica (Conexina 26)
A surdez congênita está presente em aproximadamente um em cada mil nascimentos, freqüência que pode variar de acordo com as diferentes regiões do Brasil. A origem pode ser devido a infecções no período pré e neonatal, como rubéola e toxoplasmose, bem como devido a causas genéticas. Em países desenvolvidos, onde os problemas ligados a infecções são melhor resolvidos, a surdez de origem genética está presente em aproximadamente 60% dos casos de surdez congênita.
Tem sido demonstrado que um gene localizado no braço longo do cromossoma 13, chamado de conexina 26, é o principal gene associado a surdez de origem genética e a 35delG a principal mutação presente neste gene. Através da técnica de PCR (Reação em Cadeia da Polimerase), o material genético é amplificado a partir do DNA extraido de sangue total. O resultado, após análise por eletroforese, indicará a presença ou não da mutação em um ou ambos os alelos do gene.
Importante ressaltar que a presença da mutação em um dos alelos (indivíduo heterozigoto) ou em ambos os alelos (indivíduo homozigoto) sugere que seja realizado o aconselhamento genético para a família do portador.
Em cerca de 60% dos casos de Surdez Neurosensorial Não-Sindrômica existe uma causa genética. Embora mutações em vários genes diferentes possam causar este problema na infância, o gene chamado GJB2 é o responsável por quase 50% dos casos. A deleção de uma base na posição 30 (30delG) do gene é particularmente comum e um em cada 30 indivíduos de origem européia é portador não-afetado (heterozigoto). Esta elevada frequência torna a 30delG uma das mutações mais freqüentes na espécie humana.
Assim como as demais doenças investigadas no Teste do Pezinho, a surdez, se detectada precocemente, tem maior sucesso no seu tratamento. Intervenções para reabilitação, como o uso de estímulos auditivos e de linguagem, fazem parte da terapia que demonstra melhores resultados quando iniciada nos primeiros 6 meses de vida do recém-nascido. A demora entre a suspeita e o diagnóstico da deficiência auditiva dificulta a reabilitação e pode causar prejuízos na área da comunicação e atividade social do indivíduo.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Teste genético de intolerância à lactose
Mutação C/T-13901
A lactase é uma enzima da mucosa intestinal responsável pela digestão da lactose nos seus constituintes absorvíveis, glucose e galactose. Sabe-se que sua produção persiste durante a vida adulta em algumas pessoas e em outras não. A variante genetica responsável por esta característica foi identificada em 2002. Trata-se da mutação C/T-13901 no promotor do gene da lactase. Essa mutação faz com que o gene permaneça ativo após a suspensão da lactação.
Assim, os portadores desta variação são tolerantes à lactose devido à persistência da produção da enzima que a degrada. Os indivíduos que não produzem a enzima lactase, após a suspensão da lactação, são intolerantes à lactose e apresentam sintomas, principalmente intestinais, quando ingerem leite ou outros produtos que contenham lactose.
O teste genetico para tolerância à lactose apresenta uma alta correlação com as provas funcionais. Ou seja, os portadores do genótipo CC, genótipo associado a não persistência na produção de lactase, tendem a apresentar provas funcionais alteradas. Ao realizar uma revisão sistemática dos estudos disponíveis que compararam o teste genetico com uma prova functional , chegou-se a conclusão que o teste genetico tem 79% de sensibilidade e 83% de especificidade.
O novo teste pode predizer com alta probabilidade se um indivíduo é tolerante à lactose ou não. Desta forma, o novo teste é considerado uma ferramenta importante na triagem da condição. Neste sentido, alguns protocolos sugerem que o teste genetico seja realizado antes da prova funcional, que pode até mesmo ser dispensada diante de um paciente com sintomas de intolerância e um genótipo CC. O teste genetico para tolerância à lactose está disponível no Laboratório Neurogene.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
Trombofilias hereditárias
O que é?
A trombose venosa é uma doença multifatorial definida por um conjunto de anormalidades, associadas a hiperatividade do sistema de coagulação do sangue e/ ou ocorrência de fenômenos trombóticos.
O seu aparecimento é devido tanto a fatores genéticos quanto a efeitos ambientais. Dentre os fatores ambientais podemos destacar falta de exercícios físicos, idade avançada, uso de anticoncepcionais orais, reposição homonal, período pós-operatório, pós-parto dentre outros. A trombose, entretanto, tem um forte componente genético com mutações já estabelecidas levando a um aumento na predisposição à doença. A primeira, uma mutação do gene do Fator V de Leiden, é encontrada em aproximadamente 5% da população e é responsável por 20-30% dos eventos de troboembolismo venoso.
A segunda mutação no gene da Protrombina está associada com o aumento da concentração da protrombina plasmática aumentando o risco para tromboembolismo venoso e cerebral. A terceira forma de trombofilia hereditária, a mutação do gene da Metilenotetrahidrofolato redutase-MTHFR), é a mais freqüente causando o aumento moderado de homocisteína e pode ser encontrado em 5-15% na população.
O Laboratório Neurogene oferece os exames para detecção das três mutações mais freqüentes.
Os exames são indicados para:
• Pacientes que já tiveram trombose venosa
• Pacientes com histórico familiar de trombose venosa
• Mulheres que utilizam ou desejam utilizar anticoncepcionais
• Pacientes em reposição hormonal
• Grávidas com pré-eclâmpsia
• Pacientes com trombose pós-parto
• Pacientes com perdas fetais repetidas
• Pacientes com perdas fetais repetidas de 1º trimestre
• Pacientes com perdas fetais repetidas de 2º trimestre
• Grávidas com restrição de crescimento intra- uterino
• Grávidas com deslocamento da placenta
Os testes poderão ser feitos pelo Laboratório Neurogene em conjunto (triagem) com desconto ou separadamente em DNA extraído de 1ml de sangue colhido com EDTA. O material poderá ser colhido no Laboratório Neurogene ou encaminhado a este.
1 – Teste da mutação G1691A do Fator V de Leiden
O maior fator de risco genético conhecido associado com trombose venosa é a resistência do Fator V à clivagem da proteína C ativada. Trata-se de uma alteração autossômica dominante, que interfere na atuação da proteína C, na sua forma ativada, que é um dos fatores reguladores do sistema de coagulação.
Esta resistência na maioria das vezes é causada por uma mutação no gene do fator V. Quando encontrada em homozigose, o risco de desenvolvimento de trombose venosa pode aumentar em até 90 vezes em comparação com indivíduos normais. Nos indivíduos heterozigotos o risco também é aumentado em aproximadamente oito vezes.
Metodologia: Isolamento do DNA genômico de leucócitos, seguido de amplificação por PCR, digestão enzimática e eletroforese.
2 – Teste da mutação G20210A no gene da Protrombina
A Protrombina é uma proteína precursora da Trombina, uma enzima que atua em processos procoagulantes, anticoagulantes e antifibrinolíticos do sangue. A presença da mutação G20210A no gene da protrombina eleva as concentrações de protrombina plasmática, fazendo com que os indivíduos que possuam a mutação tenham um risco 3 vezes maior de desenvolver Trombose Venosa que os indivíduos normais. Este risco aumenta significativamente pelo uso de contraceptivos orais (149 vezes) e na gravidez.
Metodologia: Isolamento do DNA genômico de leucócitos, seguido de amplificação por PCR , digestão enzimática e eletroforese.
3 – Teste da mutação C677T no gene da enzima METILENOTETRA-HIDROFOLATO REDUTASE (MTHFR)
A presença desta mutação em heterozigose não aumenta o risco de trombose. Entretanto, em homozigose a mutação leva a um aumento de homocisteína no plasma e o risco de trombose venosa aumenta 5-6 vezes. A presença de outras mutações neste gene que podem levar a um aumento da homocisteína plasmática não são detectadas neste exame.
Metodologia: Isolamento do DNA genômico de leucócitos, seguido de amplificação por PCR, digestão enzimática eletroforese.
Os testes acima poderão ser feitos pelo Laboratório Neurogene em conjunto (triagem) com desconto ou separadamente em DNA extraído de 1ml de sangue colhido com EDTA. O material poderá ser colhido no Laboratório Neurogene ou encaminhado a este.
Método: É utilizado teste molecular baseado na Reação em Cadeia da Polimerase (PCR) para o diagnóstico dos genes específicos.
Material: 5ml de sangue com EDTA
Prazo de Entrega: 30 dias
INTERPRETAÇÃO DOS TESTES
- Trombofilias e Infertilidade – Associação com perdas fetais (Informe Abril/2009)
- Pré-eclâmpsia – fatores genéticos que predispõem (Informe Abril/2009)
- Enzima Conversora da Angiotensina – associação com doenças cardiovasculares (Abril/2009)
- Predisposição à Hipertensão e Doenças Cardiovasculares (Informe Abril /2009)
- Pesquisa genética para APOE – Associação com doenças cardiovasculares e Alzheimer (Abril 2009)
- Falha Ovariana Precoce ou Menopausa Precoce – Associação com a Sindrome do XFrágil (Abril 2009)